Google's Co-Scientist paper (Gottweis et al., Nature, 2026) describes a multi-agent hypothesis-generation system: six specialized Gemini agents (Generation, Reflection, Ranking, Evolution, Proximity, Meta-review) plus a Supervisor. The team validated it wet-lab in three biomedical settings, including AML drug repurposing, liver fibrosis, anti-microbial resistance, and reported per-paper benchmarks against the SOTA reasoning models of early 2025.
This repo is an independent reimplementation. The paper's source code isn't public (?!), but the supplement contains pseudocode for each agent (reference/8 Pseudocode…) and the full prompts (reference/9 Prompts…); both were enough to rebuild the agent roster faithfully (?) What follows is a side-by-side: the design choices, the gaps relative to the paper, and what we learned from running the included co-scientist bench harness against the paper's preference-ranking comparison.
GitHub: https://github.com/Kaimen-Inc/Co-Scientist
Full Benchmark Results showcased on AI Scientist Arena
TL;DR
- The agents are re-implementation of the paper's agents. Roster, prompts, debate/tournament/evolution logic all follow the supplement. The same six worker types + Supervisor; Elo-1200 init; the same meta-review→prompt-append "learning without backprop" loop.
- The infrastructure is not the paper's infrastructure. The paper runs on Google internal scheduling; we built a durable SQLite-backed task queue with bounded concurrency, FAISS-backed proximity, and a provider-agnostic LLM layer so we can test more LLMs(Anthropic / OpenAI / Gemini / OpenRouter / Groq / Together / Mistral / Ollama / OpenAI-compat).
- What we can't reproduce: the paper's wet-lab validation (we have no lab) and the paper's compute scale (they scale test-time compute hard; we cap per-session at a few USD by default).
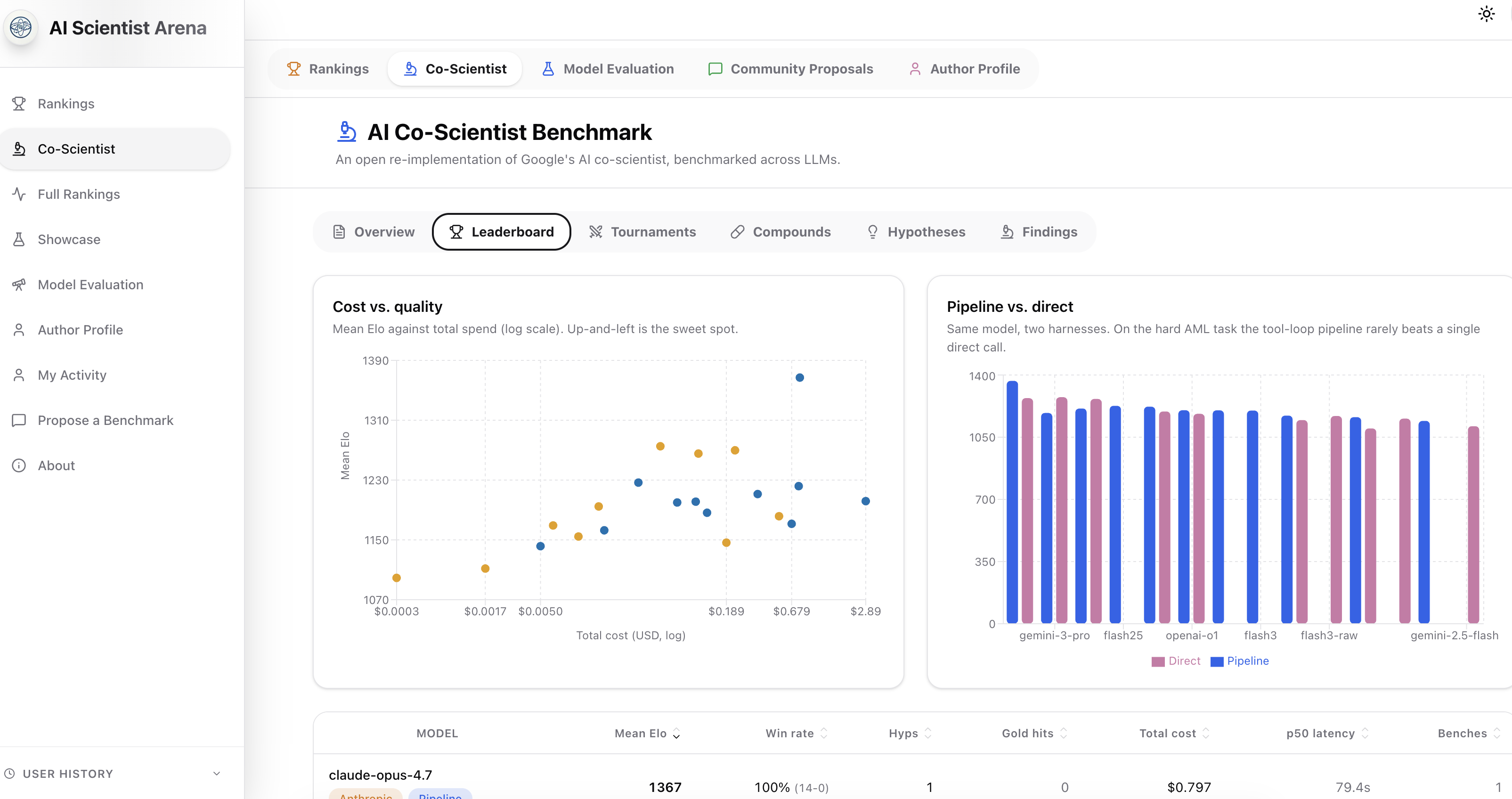
- What we can reproduce: the preference-ranking style comparison against the paper's frontier baselines. We added a wrinkle the paper doesn't: each model runs both through the full multi-agent pipeline and as a single raw LM call, in the same Elo pool, so the harness's contribution gets a number.
- What the benchmarks actually showed (20 benches, 48 AML hypotheses total): the pipeline reliably finishes and produces a hypothesis. Whether it helps the underlying model is not reproducible at n=1 — the direct→pipeline Elo delta swings sign across reruns for at least one model. Models converge on mechanisms (LSC targeting, OXPHOS, BCL-2) but diverge wildly on which drug to propose, and none of the 48 hypotheses ever hit the paper's original picks (Nanvuranlat, KIRA6, Leflunomide).
1. The architecture we kept
The agent roster, prompts, and the control flow come straight from the paper's supplement.
co-scientist run "<goal>"
│
▼
┌──────────────────────────────────────┐
│ Supervisor │ durable task queue (SQLite)
│ • parse_goal → ResearchPlan │ bounded concurrency
│ • enqueue initial Generation tasks │ lease + dead-letter + resume
│ • main loop: claim → run → follow-up│
│ • decide_next_steps when idle │ termination:
│ • finalize: meta-review overview │ BUDGET / WALL_CLOCK / ELO_STABLE / IDLE / EXTERNAL
└──────────────────────────────────────┘
│ tasks
┌─────────────────────┼─────────────────────────────┐
▼ ▼ ▼
Generation ──hyp──► Reflection ──review──► Ranking ──► Elo
literature full + pairwise vs
+ debate verification debate
▲ ▲ │
│ │ informative pairings ▼
Evolution ◄──── Meta-review ─────────────► Proximity
combine/ feedback FAISS embed
simplify/ + final + cluster /
feasibility/ overview dedup
out_of_box
The agent mapping
Paper agent
Our implementation
Notes
Generation
co_scientist/agents/generation.py
Two modes per the paper:
literature (web search → synthesize → propose) and
debate (simulated multi-turn scientific debate). Forces a
record_hypothesis tool call to make output parseable across providers.
Reflection
co_scientist/agents/reflection.py
Three review modes:
review (novelty + correctness + testability + safety, with web search),
verification (deep decomposition into sub-assumptions),
observation (does this hypothesis explain long-tail observations?). Skips the paper's "initial review" — the paper itself notes it's mostly a cost-saving filter; with our budgets it's not a bottleneck.
Ranking
co_scientist/agents/ranking.py
Elo-1200 init; multi-turn
debate for top-ranked pairs, single-turn
pairwise for the rest. Structured verdict via a forced
record_verdict tool call (the paper parses text — fragile across providers; we picked the cleaner route).
Evolution
co_scientist/agents/evolution.py
Four strategies (the paper lists six; we collapsed "inspiration from existing" + "enhancement through grounding" into our four since they share the same prompt scaffolding):
combine,
simplify,
feasibility,
out_of_box. Generates new hypotheses, never mutates existing — same as the paper, to protect tournament-tested entries.
Proximity
co_scientist/agents/proximity.py
FAISS
IndexFlatIP per session; Voyage → OpenAI → hash-fallback embedder chain. Drives dedup and informative tournament pairings (closer pairs get scheduled more often).
Meta-review
co_scientist/agents/metareview.py
Two modes:
system (synthesize patterns from reviews + match transcripts, feed back as appended prompt to all agents — the paper's "learning without backprop"),
final (research overview at end of run).
Supervisor
co_scientist/agents/supervisor.py
Parses goal → research plan, schedules worker tasks, computes summary stats periodically (Elo distribution, queue depth, agent effectiveness), decides what to schedule next, decides when to stop.
The prompts
The 14 Jinja templates in config/prompts/ are direct ports of the paper's supplementary prompts. We kept the structure verbatim (modulo Jinja interpolation for goal, hypothesis, prior context). When the paper publishes prompts in reference/9 Prompts for the specialized agents in .md, that file is the canonical reference.
Termination
The paper terminates when summary statistics suggest a "terminal state." We made that explicit — five reasons:
BUDGET — token/USD cap hit (default $2 per run)WALL_CLOCK — deadline crossedELO_STABLE — top-K Elo hasn't moved more than ε over the last N matchesIDLE — empty queue + no follow-ups to scheduleEXTERNAL — user invoked pause/abort
Live in co_scientist/orchestrator/termination.py.
2. What's not the paper
Three categories.
2a. Infrastructure: we couldn't reuse theirs
Their async task framework runs on Google internal infra. Ours runs on:
- SQLite + WAL with
busy_timeout and an idempotent migration runner. 15 tables: sessions, hypotheses, reviews, tournament_matches, elo_journal, tasks (durable queue), transcripts, system_feedback, embeddings_meta, spans, events, bench_runs, bench_candidates, bench_matches.
- A lease/dead-letter task queue with resume after crash. The paper's framework restarts cleanly from context memory; ours restarts cleanly from
tasks.status = 'leased' AND lease_expires_at < now().
- An event bus that fans out to SSE for the live web UI (
co_scientist/web/).
- A provider-agnostic LLM layer (
co_scientist/llm/) — the paper is Gemini-only; we support 9 provider backends and let you pin a different model per agent role (generation, reflection, ranking_pairwise, metareview_final, etc.) via TOML. Prompt-cache breakpoints are wired up for Anthropic; reasoning effort knobs for OpenAI o-series and Anthropic thinking.
2b. Validation: we couldn't run wet lab
The paper's three real-world validations (AML drug repurposing in vitro, liver fibrosis epigenetic targets in hepatic organoids, recapitulating the cf-PICI mechanism) require a lab. We have none.
The closest in-silico proxy we built is recall against curated answer keys from the paper:
label
size
what it is
aml-repurposing-paper-top3
3
The paper's
strict methodology picks — no prior preclinical evidence, no external inputs (no DepMap, no expert curation).
Nanvuranlat, KIRA6, Leflunomide.
aml-repurposing-paper-5
5
The broader 5-drug list from the main text.
Binimetinib, Pacritinib, Cerivastatin, Pravastatin, Dimethyl fumarate.
The matcher checks whole-token, case-insensitive against every searched field (title, summary, full text, entities, citation excerpts) of every hypothesis a candidate produces. Drug class mentions (e.g. "DHODH inhibitor") don't count — the name has to be there.
2c. Scale: we couldn't run their compute
Their Fig. 2a is over 203 research goals with the system left to scale test-time compute. Their Fig. 2b is 15 expert-curated goals each run head-to-head against five other LLMs. Our default budget is $2/session and --n 1; the full picture is in BENCH_RESULTS.md.
3. The bench harness — a knob the paper doesn't have
The paper compares Co-Scientist against frontier LLMs (Gemini 2 Pro/Flash, o1, o3-mini-high, DeepSeek R1) via Elo. We do the same thing — --preset paper reproduces those baselines via OpenRouter. But the paper's comparison answers "is the system better than the underlying model?" by comparing Co-Scientist (on Gemini 2.0) against plain Gemini 2.0, plain o1, etc. The harness's contribution is conflated with everything else.
Our *-vs-raw presets cleave it cleanly: same model, two modes, same Elo pool.
pipeline — model runs through the full Generation agent (literature tools + tool-use loop + dedup + forced record_hypothesis)direct — same model, same prompt, single LM call with the same forced record_hypothesis, no tools
Round-robin pairings, structured verdict from one fixed judge model (so no candidate scores its own work), all matches recorded with rationale in bench_matches.
This is the only knob in the paper's space that we're set up to measure cleanly. So we did, twice, and the result is — instructively — noisier than I expected.
4. What 20 benches actually showed
Full table is in BENCH_RESULTS.md. The summary:
4a. The harness completes; whether it helps doesn't replicate at n=1
After the d207233 pipeline-reliability fixes, every candidate finishes pipeline mode in the recent runs. The misses are external — a transient HTTP 429 and gemini-2.5-pro intermittently returning an empty completion on the forced final call (2 of 3 attempts). So "the harness ships a hypothesis" is reliable.
The Δ-Elo from direct to pipeline is not reliable across reruns. Same preset, identical settings, two runs:
model
run 1 Δ Elo
run 2 Δ Elo
claude-haiku-4.5
+180 (raw 1-9, pipe 10-0)
−28 (raw 10-2, pipe 8-4)
openai-o1
+43
+29
Haiku's raw win-loss alone flipped 1-9 → 10-2 across the two runs. With one hypothesis per candidate and ~2 matches per pair, the tournament gets dominated by which single hypothesis happened to get sampled, not by the mode. The earlier-draft headline "pipeline beats raw" was reading sampling noise.
For single-run deltas, the spread is wide and doesn't line up by provider:
model
Δ Elo (single run)
claude-opus-4.7
+97
gemini-2.5-flash
+172
gemini-2.5-pro
+47
gpt-5
+26
gemini-2.0-flash
−48
gemini-3-flash
−36
gemini-3-pro
−89
Within Google alone the 2.5 models gain (+172, +47) and the 3.x models lose (−36, −89). The only signal we'd call repeatable is openai-o1, modestly ahead in pipeline mode across both reruns.
Practical implication: at this --n, don't read a single bench's Elo as a model verdict. To put a number on harness contribution per model needs many more seeds (higher --n, more --matches) so the per-hypothesis variance washes out. The paper sidesteps this entirely by running on 203 goals and 15 goals respectively; our $-budgeted runs can't.
4b. Models converge on mechanisms, diverge on drugs
Across all 48 AML hypotheses recorded on this codebase (every bench × every candidate × every mode):
recurring theme
hypotheses (of 48)
leukemic-stem-cell (LSC) targeting
28
OXPHOS / mitochondrial complex I
8
BCL-2 / MCL-1 (Venetoclax axis)
7
FLT3-ITD
6
fatty-acid oxidation
5
ferroptosis
3
These are the well-known AML vulnerability classes. The agreement is not surprising.
At the drug level it's a long tail of one-offs. Compounds proposed more than once:
- Itraconazole ×5 (as an OXPHOS inhibitor)
- Auranofin ×2 (thioredoxin-reductase)
- Venetoclax ×6 — but always as the resistance/combo context, not the novel candidate
Every one of those three already has prior published AML evidence. The strict prompt explicitly forbids that. The models default to the familiar; "recurrence across models" is a weak novelty signal at best.
4c. None of 48 hypotheses hit the paper's original picks
0/3 on aml-repurposing-paper-top3 (Nanvuranlat, KIRA6, Leflunomide).
0/5 on aml-repurposing-paper-5 (Binimetinib, Pacritinib, Cerivastatin, Pravastatin, DMF).
The proposed drugs by our co-scientist include: bazedoxifene (GP130/IL-6), pirfenidone (TGF-β/p38), meldonium (carnitine depletion), pitavastatin (isoprenylation), brensocatib (DPP1), sitagliptin (DPP4), niclosamide (STAT3), denifanstat (FASN). Several may be pre-clinically interesting. They're just not the Nature paper's picks. With --n 1, the match is mostly a draw from a wide distribution; the paper achieves hits by running massive tournaments with iterative refinement until top-ranked hypotheses converge — that's the part our $2-per-session budget can't afford.
5. What this exercise tells us about the paper
A few things the reimplementation makes more visible:
The architecture is portable. Stripping the Gemini-only assumption out wasn't hard — the agent prompts are mostly about thinking style (debate, deep verification, out-of-box evolution) and the LLM is a substrate. Any tool-using LM with reasonable context length plugs in.
The Elo tournament is doing some work for some modlels. It's the substrate that turns single sampled hypotheses (high variance) into a stable ranking (lower variance). At our scale the tournament is too thin to stabilize for all models, but may not improve all models (like gemini 3 series), pending more testing.
What is the gold standard? Even strong models hit 0/3 on the strict top-3 list proposed by the original paper, because "find the same novel drug a paper found" is a sample-of-one match against a high-cardinality distribution. The paper avoided this by generating thousands of candidates and ranking, then letting expert oncologists pick the top 30 → 5. Mechanism-class recall (LSC, OXPHOS, fatty-acid oxidation) seems to be a better fit and it may have hold up. Cancer has been cured in vitro many times, whether these are actually good drugs remained to be tested...
6. Where to look in this repo
README.md — install, run, configuredocs/BENCH_RESULTS.md — auto-generated, every bench's per-candidate Elo, every hypothesis, original paper hits, file pointersco_scientist/agents/ — the seven agentsconfig/prompts/ — the 14 Jinja prompts derived from the paper's supplementreference/ — paper source materials (pseudocode, prompts, diagrams), gitignoredco_scientist/bench/ — the head-to-head harness, original paper hit scorer, presetsscripts/build_bench_report.py — regenerate BENCH_RESULTS.md from SQLite after a new bench
Apache-2.0. Independent reimplementation. I'm not affiliated with Google or the paper's authors & thank them for the initial contribution.
GitHub: https://github.com/Kaimen-Inc/Co-Scientist
 KH
KH